本文内容仅可为解决实际问题时提供一个参考,不作为2010版GMP的依据或判定原则。
1、针剂在线灭菌时:
A)呼吸过滤器如何保证达到无菌保证要求?
B)滤芯在灭菌浸湿后,通气效果变差,如何处理有效?
解答:
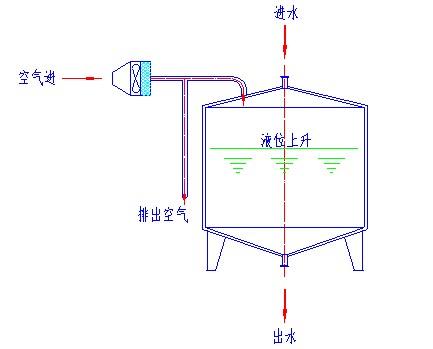
呼吸器过滤器是可以作SIP或离线灭菌的,它的要求取决所使用的目的。如纯化水贮罐上用的呼吸器过滤器可采用离线灭菌法(没有必要设在线灭菌),也可采用下图所示高效过滤器主动送风的形式,在任何情况下保持贮罐的正压状态。
冻干及其它无菌生产设备上的疏水性过滤器的灭菌,通常采用电加热或压缩空气吹干的方法使其保持运行状态,但是不同的情况有不同的处理方式,具体可以按供货商规定的要求处理。
2、压缩空气系统确认中测定压缩空气中水份的含量(使用德尔格压空仪测定)水份含量指标换算后即可代表露点值?该含量可接受标准应如何考虑,是否不同洁净级别之间水份要求不同?无油压缩机是否可不测试含油量?
解答:
压缩空气的控制标准是结合所需生产的药品特征而设计控制标准的,有露点、微粒、含油量、微生物限度等。压缩与生产区的洁净级别没有多大直接相关性。压缩空气有国标,企业应根据产品情况制订本企业的标准,原则上说,它不得影响产品的工艺及质量。
因为现空气的污染情况比较严重,无油空气压缩机并不可能解决“空气污染问题”,采用无油空气压缩机时仍应对含油量控制。
3、压缩空气应确认哪些项目?现场检查中要检哪些指标?如何检查?
解答:
在制药企业中,压缩空气常见的二种用处分别是:气动阀的动力源;生产工艺用气体,例如洗瓶的吹干、灭菌柜的补气(特别是灭菌时作软包装的背压)以及配制罐压药液等等。动力源通常只有压力的要求,但工艺用气体要考虑产品的特点,如是无菌分装,对干燥的要求可能会高一点(控制露点);如是液体产品,则不需要特别干燥,因此,GMP规范不规定技术要求,检查方法请见国标。注意,不要离开了产品的工艺要求,去提露点(干燥度)、微粒等限度标准。
4、过滤系统终端过滤芯的完整性测试试验一定需要在线监测吗?而压缩空气系统、制氮系统的滤芯如何测试和清洁?
解答:
GMP并没对此规定,有条件时建议在线监测,但这不是强制要求。与液体除菌过滤器不同,压缩空气系统、制氮系统除菌过滤器的滤芯可按供货商提供的方法进行测试,定期检查;其前道其它用处的过滤器(例如去除油雾),均应按工艺要求及供货商要求处理。
5、大输液生产过程中罐装及管路的批次清场有何要求?批与批之间必须在线清洗吗?
解答:
这是一个比较现实的问题,清洗的目的是什么,要解决什么问题?因为清洗后,贮罐及管路等会有部分积水,除非你用压缩空气或氮去吹干,否则会影响下批初始产品的浓度,清洗需要操作时间,也会造成浪费。因此,如是批与批(同一产品、相同浓度及包装规格)的连续生产,可能不作清洗,也可能只需要简单的清洗(只清洗贮罐),这要看产品是否容易分解、长菌或其它对产品质量可能的不良后果。换言之,应根据风险评估的结果作出决定。

6、灭菌冷却过程中为了维持瓶内外压力平衡,需补充压缩空气,请问此使用点的压缩空气是否需要除菌过滤?
解答:为了避免已灭菌产品被再次污染,补充的压缩空气应经除菌过滤。
7、用于塑料瓶气洗的终端压缩空气滤器完整性实验如何做?怎样才能有好的办法检测到滤芯漏了?
解答:
新版GMP附录1无菌药品第四十二条进入无菌生产区的生产用气体(如压缩空气、氮气,但不包括可燃性气体)均应经过除菌过滤,应当定期检查除菌过滤器和呼吸过滤器的完整性。请注意,气体过滤器的截留效率比液体过滤器高出10~100倍。国际上,将气体过滤器分为3类:
◇ 直接与无菌产品接触(如:无菌灌装中使用的压缩空气)
◇ 不直接与无菌产品接触(如:发酵过程通气过滤器)
◇ 降低微生物负荷(微生物污染水平)(如:HVAC中的HEPA高效过滤器)
所提塑料瓶是用于无菌药品还是非无菌药品生产不明确,这里分别说明一下。如果是无菌生产(如冻干),冻干腔室和工艺用气体除菌过滤器是做在线灭菌的,但通常不是每批进行在线灭菌,其完整性也不每批检测,而是定期测试,这样做的风险是--如后一次测试证明除菌过滤器确实损坏,则从上次测试合格后生产到这次测试期间的产品将作无菌不合格处理。为了避免经济的风险,可购买在线完整性测试仪进行测试;如是最终灭菌产品,过滤器实际上可长期使用,定期检查,例如这种检查可以是每半年、一年检查一次,由企业根据实际使用情况及结果进行风险分析,确定检查周期。
8、公用系统(如压缩空气系统、制氮系统)其管道是否需要定期消毒?如何做?
解答:
GMP对此无技术性规定,应按供货商的维修要求处理,管路的清洁应在安装阶段完成。用作动力源的压缩空气不需要消毒。工艺用压缩空气系统、制氮系统的管路,用于最终灭菌产品时,也不要求消毒;当于不可最终灭菌的产品(已除菌过滤后的药液)接触时,部分管路(软管)与过滤器是经灭菌的。
9、与药液直接接触的压缩空气或氮气在系统安装时不进行全面的验证,日常监测是否可只监测含菌量及粒子?
解答:
公用介质通常采用确认,而不称验证。与药液直接接触的介质系统均应确认,未经确认的系统不应投入使用。值得强调的是,含油量是常见的确认项目,应列入确认方案,并在确认中进行确认。
压缩空气和氮均有国家及ISO/国际标准,网上可查得到标准的具体要求。不同产品对这类介质的要求不尽相同,因此,标准由企业根据产品来定,GMP对这类介质无技术性规定。另外,用于无菌生产(例如冻干粉针)的压缩空气或氮,均应通过除菌过滤。
10、工艺用压缩空气管道系统定期消毒的可操作性?能否理解:选用的无油空压--经过干燥--三级过滤--确认后在和物料接触前端再加0.2um过滤后,管道系统定期消毒,只要对压缩空气品质监测评估即可?
解答:
所提到的处理方法基本都是这样,但要根据风险来讨论要求。经处理的压缩空气进生产区时,可能有二种情况,一种是用作动力源(平时不列入监控范围);另一种是工艺或动力二者的结合(因简化管路而不将管路分得过细)。只有无菌药品的生产中,在分管路上才安装除菌过滤器,保证直接与药品或内包材接触的气体符合工艺要求,需要做的是定期检查除菌过滤器的完好性。工艺用压缩空气要检查含油量和微生物项目。
用于无菌生产(全无菌操作)的压缩空气,如冻干机平衡压力用的压缩空气,或用于压无菌药液的压缩空气等,过滤器与容器往往是做在线灭菌的,或部分软管与过滤器一起灭菌。