
【重要性】合规软件只有审计追踪和电子签名还远远不够
时间:2023-03-22 阅读:268
自2003年美国食品与药品管理局出台了联邦法规第21章 第11款(FDA 21 CFR Part 11),对电子数据的真实性、完整性和可靠性,以及电子签名有效性,收集和分析数据的软件必须是经过验证提出了要求。

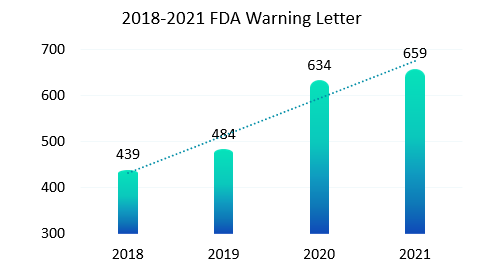
与纸质记录相比,电子记录是动态记录必然会暴露出更多的不合规操作, 2015年警告信FDA 483表格和现场观察报告频频出现,全球各地监管机构再次更新和完善了一轮法规要求。新法规施行后(2018版FDA 21CFR Part11),我们看到如下图统计,最近几年FDA发出的警告信也呈现出逐年上升趋势。作为合规软件的供应厂商,我们也明显感受到国内客户对于合规的要求也是越来越高。
每年年底/年初,制药公司、生物技术公司、CRO、CDMO等都会进行合规的企业内部审计或国家审计局委派当地审计局进行审计。今年接到多次电话咨询,无论内审还是外审都提到了操作员登录软件是否会被记录到审计追踪中。从审计关注点我们可以看出,国内对审计追踪记录完整性要求在提高,审计追踪不仅仅记录修改,还要记录登录/登出和查看信息;这也从侧面反映法规对数据的隐私性越来越重视了。(可以放钱君娣上一篇关于审计追踪的链接)。
Molecular Devices公司SoftMax Pro 7.1.2 GxP软件除了能够记录完整的审计追踪,还能够记录数据文件的生命周期,实现对数据来源和去向的全流程管理,真正做到无纸化、全流程电子记录和电子签名,最大限度保证数据记录的真实性、完整性和可靠性。
为什么我们要对数据进行全流程管理?因为2020年12月1日正式施行的国家药品监督管理局发布的药品记录与数据管理要求(试行)第二章基本要求中有如下要求,

针对这项要求SoftMax Pro 7.1.2 GxP软件新增了数据文件状态显示(如下图),在数据库中和打开数据时都会显示当前文件所处的生命周期状态。
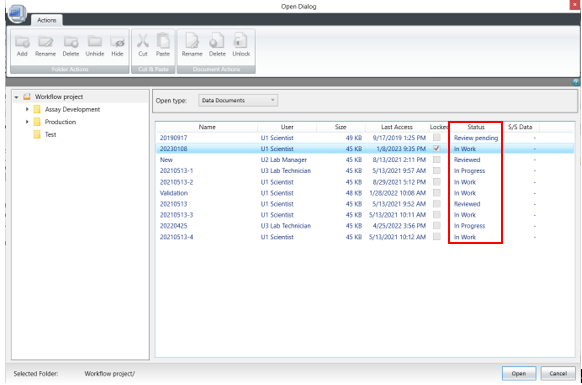
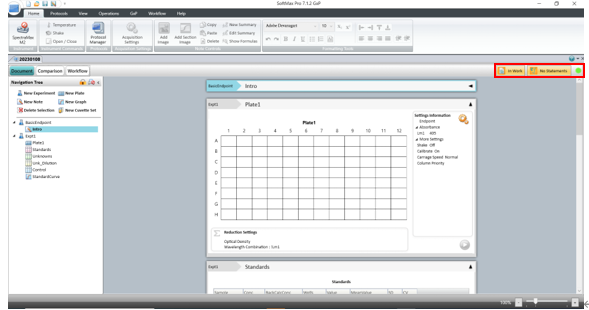
如Scientist权限新建数据状态是in work表示正在进行模板方法开发,经过Lab Manager层层审批并电子签名后可以更改文件状态并将开发好的模板release出来,这时Lab Technician才有权限打开模板进行读板和数据分析,签名后数据将被锁定不可更改,Lab Manager审核后电子签名Approved。如果发现数据有问题,可以canceled;如果因为改进流程更新方法模板,可以将更改旧模板状态成outdated,并不赋予Lab Technician打开outdated权限,以免使用错误模板读板产生不合规的数据。
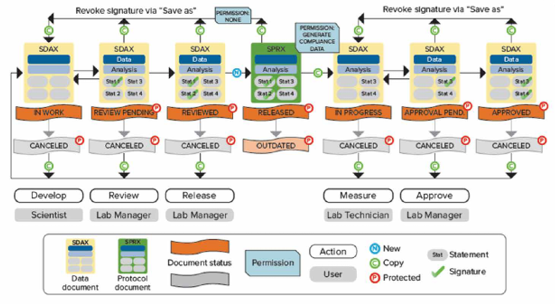
数据文件存储在SQL Server数据库中,文件的导入、重命名、移动、删除、归档等都会记录在审计追踪中,真正做到了从数据来源到最终归档全流程管控。